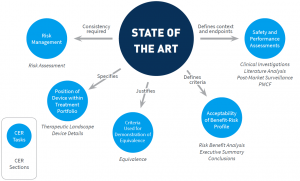
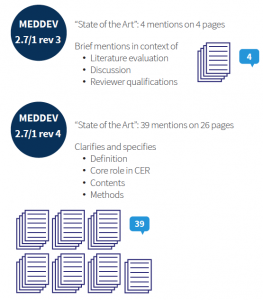
 Prior to MEDDEV 2.7/1 rev 4, guidance referred to state of the art only vaguely. MEDDEV 2.7/1 rev 3 requested the discussion of clinical data “in comparison with” and “taking account of” state of the art, and that the clinical literature data cited “reflect current medical practice and the generally acknowledged state of the art technologies”. From a methodological standpoint, this is not much guidance. Thus, methodology, depth, and presentation of the medical background for a device was largely left to one’s interpretation, and thus conducted inconsistently, or, at least, heterogeneously.
Prior to MEDDEV 2.7/1 rev 4, guidance referred to state of the art only vaguely. MEDDEV 2.7/1 rev 3 requested the discussion of clinical data “in comparison with” and “taking account of” state of the art, and that the clinical literature data cited “reflect current medical practice and the generally acknowledged state of the art technologies”. From a methodological standpoint, this is not much guidance. Thus, methodology, depth, and presentation of the medical background for a device was largely left to one’s interpretation, and thus conducted inconsistently, or, at least, heterogeneously.“Given that 3D printing is used for making device implants, many would expect tight regulation. But the EU Medical Device Regulation does not specifically regulate it, and there may be easier routes to compliance than companies think, according to attorney Joerg Schickert.”
This website uses cookies so that we can provide you with the best user experience possible. Cookie information is stored in your browser and performs functions such as recognising you when you return to our website and helping our team to understand which sections of the website you find most interesting and useful.
Read more about our privacy policy here.