The increasing number and complexity of medical devices with several functions prompted FDA to provide medical device manufacturers with premarket and postmarket regulatory guidance. In a nutshell, FDA states that it shall not regulate non-device functions of a product with multiple functions – but can consider the impact of non-device functions on the device functions. The guidance identifies the principles and premarket review practices, postmarket requirements, and multiple examples of the application of the policy.
The term “multiple function” device was first officially addressed by FDA in the 21st Century Cures Act Section 3060 that amended the Federal Food, Drug, and Cosmetic Act (FD&C Act). The guidance amended the definition of “device” removing certain software functions (medical and decision software support) and clarifies which software functions (analyze or interpret medical data) remain under FDA’s oversight.
The guidance document is intended to clarify FDA’s policy approach to all multiple function devices, considerations for the design and risks of multiple function device products, and impact assessment of non-device software functions or “other functions” on safety and effectiveness. Furthermore, the guidance provides recommendations on the premarket submission content FDA expects for a device function that is part of a multiple device product. Modifications and postmarket requirements are also addressed.
The policy is risk based with a functionally focused approach to software that is platform independent. The regulation is narrowly tailored to the device’s risk based on the function with the ultimate intent to protect patient safety.
In the guidance, FDA clearly delineates “other functions” and device function-under review.
Definitions
To begin, FDA defines function as a “distinct purpose of the product” and further clarifies that function could be the intended use or a subset of the intended use of the product. For example, a device with the intended use of storing, transferring, and analyzing data has three functions while a device with the intended use of analyzing data has only one.
“Other function” is considered a function that:
Does not meet the definition of device;
Meets the definition of device, but is not subject to premarket review (e.g., 510(k)-exempt); or
Meets the definition of device, but for which FDA has expressed its intent not to enforce compliance with applicable regulatory controls.
Multiple function devices are comprised of functions that are subject to FDA’s regulatory oversight and “other functions” that are not.
Device function-under review is defined as a function for which FDA is conducting premarket review.
Policy: Premarket Review Considerations for Multiple Function Device Products
The guidance clarifies that while “other functions” that are part of a multiple-function device are not the subject of FDA review, FDA may assess the impact of “other functions”when conducting the premarket review to determine the devices safety and effectiveness.
“Other Functions”Should be Separated in Design and Implementation
FDA considers the separation of functions of the device-under-review from “other functions” important for risk control. Ideally, the separation occurs early in the design with a higher degree of separation allowing for easier review of the independent safety and effectiveness of the devices function.
Making architecture decisions early in the design cycle that facilitate risk control especially allow for the evaluation of optimal separation and support segregation of:
Logic
Architecture
Code
Data partitioning
This separation is considered “especially important when considering cyber security risk and mitigations.”
Risk Analysis Needs to Considerthe Impact of Other Functions
The guidance outlines the following as critical for manufacturers to consider regarding the impact of other functions.
Role of the “other function” in the devices function-under-review’s performance;
Limitations of the device function-under-review when using the “other function:”
Developing appropriate hardware and software resources specifications(s) for the product with multiple functions to ensure minimal impact of the “other function” on the performance of the device functions-under-review;
How to ensure appropriate actions are taken by the end user when using the device function-under-review with the “other functions;” and
Identification, evaluation, and mitigation of any additional risks, including cybersecurity risks, in the device-functions-under-review when used in combination with the “other function.”
When separation cannot be achieved, the interconnectedness and interdependencies of the devices function and “other function” needs to be included in the hazard analysis. Furthermore, an appropriate control must be created.
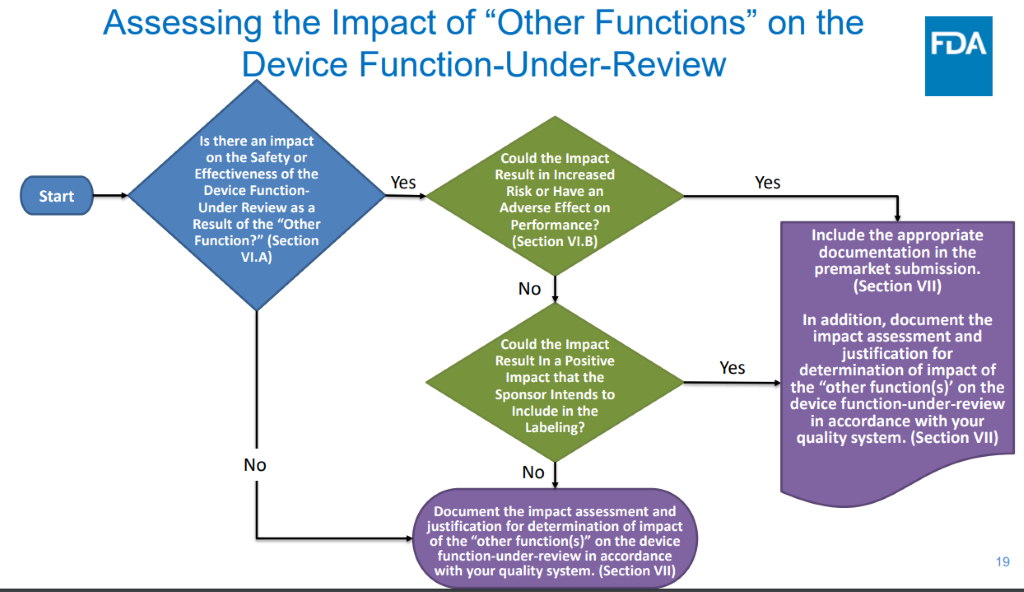
Assessing the Impact of “Other Functions” on the Device Function-Under-Review
FDA is clear on how it intends to assess the impact of “other functions” and provides a flow chart to guide manufacturers through the assessment.
Manufacturers need to conduct a risk assessment to determine if the “other functions” impact the safety and effectiveness of the device under review. The other functions could act to have a positive effect or an adverse effect.
A two-step process summarizes the premarket assessment.
First, is there an impact on the Safety and Effectiveness of the device function-under review as a result of the “other function?” If yes, could this be a negative impact, meaning an increased risk or an adverse effect on performance of the device function under review?
Positive impact assessments must include the beneficial impact of “other functions” on the device-under-review when the device is operating as intended. In addition, the manufacturer must confirm that the “other functions” do not impart an adverse impact if the device-under-review fails to operate as intended.
Negative impact assessments must identify the potential for an increased risk and/or adverse effect on performance “due to the combination of the other function with the device-under-review.
“Other functions” that impact the device under review must be evaluated and included in the hazard analysis.
Manufacturers need only added other functions to the submission if the positive effects are represented on the device label – a labeled positive impact – or if it may negatively impact the device that is under review.
Examples of Multiple Function Devices
The FDA provides 7 examples of multiple function device products in Appendix 1. The examples provide a description of the product, product functions (see list below), description of the impact of the “other function” on the device function-under-review and an explanation of the impact assessment.
Below is a list of the hypothetical example device products. See the guidance for details on impact assessment.
Skin cancer detection software application
Device function under review:
Software app that detects skin cancer
“Other function:”
Smart phone computing platform
Camera on the computing platform
Hand-held coagulation device
Device function under review:
Hand help coagulation instrument
Coagulation (prothrombin time) test
“Other function:”
Docking station
Interface to transmit the data to the HIS
Traumatic brain injury determination
Device function under review:
Collection and recording of EEG signals
Analyzing EEG signals and diagnosing TBI
Presentation of results
“Other function:”
General use computing platform
Electronic administration of questionnaire
Pain treatment app
Device function under review:
Electronic Nerve Stimulation as a treatment for pain
App used to control the level of stimulation
“Other function:”
Mobil platform Bluetooth transceiver and connectivity
Transmission of vital sign measures to an Electronic Health Record (HER)
Device function under review:
Vital signs acquisition
“Other function:”
Transmission software for sending data to the HER system
Wi-Fi card
Energy-delivering device with optional app (MDDS)
Device function under review:
Energy-delivering aesthetic device
“Other function:”
Mobil app that integrates with device and transfers treatment parameter data (e.g., number of treatments, treatment parameters) to a cloud-based storage – no real-time transmission is allowed
Smart phone computing platform
Pulsed ultrasound and biopsy needle guide kit
Device function under review:
General purpose diagnostic ultrasound system
Biopsy needle tracking functionality
“Other function:”
510(k)-exempt needle guide kit
Postmarket Guidance
The guidance applies to post-market regulations and implicitly states that device functions must comply with design control requirements and expectations surrounding adverse event reporting.
FDA reinforced its position that it does not intend to “enforce general control requirements for device functions for which FDA has expressed its intention to not enforce applicable regulatory controls at this time.”
Multiple function device products that have undergone modifications of the “other function” must be assessed to determine if the change has the potential to significantly impact the safety or effectiveness of the device function that was the subject of FDA review.
This guidance document was written to clarify current regulations as well as to promote innovation and patient engagement with digital health products.
The generation of technical documents that are required for medical device premarket approval requires time and expertise. Consider increases in your company’s efficiencies by working with a team of medical writers that are document experts. At Criterion Edge, we take pride in teaming up with industry experts to provide high quality support. Please reach out to us for a free consult.
Fill out this form, or contact us at info@criterionedge.com, and we would be happy to answer any of your questions as well as book you an appointment to discuss your project needs during a time that works for you.
Are You Reg. Ready? Get your medical affairs team primed to manage new regulatory requirements
In this discussion, we provide an overview of how to plan and coordinate change within an organization to meet upcoming EU IVDR regulatory requirements. We will be highlighting key steps involved in helping medical affairs transition to a more rigorous regulatory environments, and how to evaluate talent gaps, team leadership composition, and process challenges of your product portfolio.
Driving Innovation to Success in the Market: Strategic Considerations
In this discussion, our panel of proven leaders discuss the key elements that support and propel the innovation process in the medical device, pharmaceutical, and IVD industries. We will be highlighting key areas of the process, important players in the pathway to the market, and how successful innovations spawn new innovations in new markets such as the digital health space, and more.
Privacy Overview
This website uses cookies so that we can provide you with the best user experience possible. Cookie information is stored in your browser and performs functions such as recognising you when you return to our website and helping our team to understand which sections of the website you find most interesting and useful.
Strictly Necessary Cookie should be enabled at all times so that we can save your preferences for cookie settings.
If you disable this cookie, we will not be able to save your preferences. This means that every time you visit this website you will need to enable or disable cookies again.
3rd Party Cookies
This website uses Google Tag Manager and Pardot's tracking features to collect information such as the number of visitors to the site, and the most popular pages. Keeping this cookie enabled helps us to improve our website.
Please enable Strictly Necessary Cookies first so that we can save your preferences!