In vitro diagnostic (IVD) medical devices in the European Union are being held to a higher standard of scrutiny with the introduction of In Vitro Diagnostic Regulation (IVDR) 2017/746 by the European Commission (EC).
Overview of In Vitro Diagnostic Regulation 2017/746
The implementation of IVDR dramatically changes the regulatory landscape for IVD medical devices. Some of the biggest changes include an expanded definition of IVDs, a new classification of IVDs, the requirement of Unique Device Identification (UDI) on each device, an expansion of the Quality Management System, and the increased need for Notified Body (NB) review.
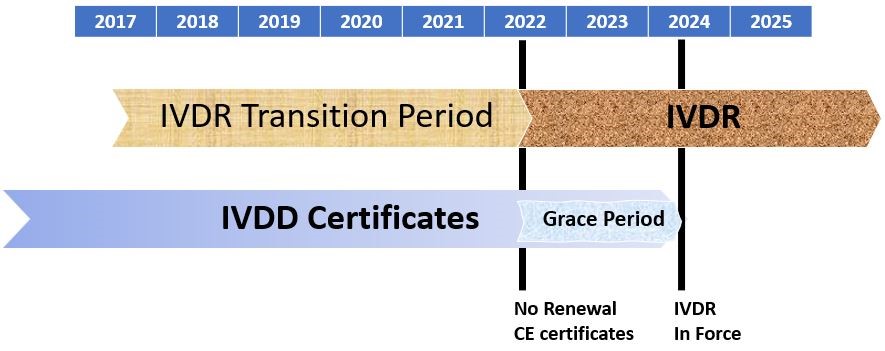
The transition into IVDR is already underway. IVDs marketed in the EU will continue to require a CE Marking certificate to verify that the device meets all the regulatory requirements. Failure to meet the IVDR deadlines could be very costly to manufacturers that would either lose their CE Marking or fail to obtain one. The timeline below outlines the deadlines for manufacturers to become compliant with IVDR 2017/746.
IVDR Timeline
The IVDR is being implemented over a 5-year transition period in which it will fully replace the current In Vitro Diagnostic Directive (IVDD) 98/78/EC (Figure 1). IVDR 2017/746 was published May 5th, 2017, in the Official Journal of the European Union, and officially went in to affect May 25th, 2017; that means we are almost halfway through the transition period. During the transitional provisions, manufacturers can meet either IVDD or IVDR requirements. However, starting May 26, 2022, all new IVDs must go through IVDR. Some devices that lawfully obtained IVDD certificates have another 2-year grace period and may continue to be made available until May 27, 2025 (depending on serval factors).
Article 110 Transitional provisions state that “Certificates issued by notified bodies in accordance with Directive 98/79/ED[EC] prior to 25 May 2017 shall become void by 27 May 2024.” IVDs that obtained certificates in accordance with IVDD prior to May 25, 2017 are valid until the end of the period indicated on the certificate, with some exceptions. Certificates issued under Directive 98/79/EC Annex VI become void at the latest May 27, 2024. If a device is lawfully placed on the market under IVDD prior to May 26, 2022 and placed on the market from May 26, 2022 by a certificate (refer to paragraph 2 and 4), the IVD can be on the market or put into service until May 27, 2025.
See IVDR Article 110 and the IVDR Corrigendum number 8 for specific details.
Starting May 26, 2024 IVDR will be full application, and all IVDs must be fully compliant.
Figure 1. IVDR Timeline
It may seem that there is plenty of time to obtain IVDR compliance, but there are many hurdles to obtaining CE Marking under this new regulation. Also consider that there are major changes in almost every aspect regulation for medical devices in the EU, including MDR, MedDev2.7/1 rev.4, and CERs.
Scope of IVDR
The scope of IVDR is massive, and it impacts all aspects of in vitro diagnostic device manufacturing.
The definition of an IVD is expanded to included software and companion diagnostics.
This requires IVDs to use a new device classification system that will place 70%-80% of IVDs in a new category, and, thus, requiring significant work to obtain or maintain market approval.
There is no grandfathering of any device. Even those currently on the market, must conform to the new IVDR standards.
If you are marketing or intend to market any type of In Vitro Diagnostic Medical Device in the European Union, it is time to act. With the need for NBs substantially increasing, it is important to start the process of performing a Gap analysis and finding a NB.
Let’s start by first looking at the newly expanded definition of IVDs under IVDR 2017/746.
If you are marketing or intend to market any type of In Vitro Diagnostic Medical Device in the European Union, it is time to act. With the need for NBs substantially increasing, it is important to start the process of performing a Gap analysis and finding a NB.
Let’s start by first looking at the newly expanded definition of IVDs under IVDR 2017/746.
Definition of In Vitro Diagnostic Devices
Section 1, Article 2 of IVDR 2017/746 expands the definition of IVDs. The addition of software and companion diagnostics to the definition of in vitro diagnostic devices significantly expands the definition of IVDs.Software is a medical device according to the definition of IVD if that is its intended purpose; thus, software as part of an instrument, software as a medical device, and apps are included in the definition of IVDs and fall under IVDR regulation. This includes genetic testing, companion diagnostics, as well as stand-alone software.
The specific definition of In Vitro Diagnostic devices from IVDR 2017/746 is below.
(2) ‘in vitro diagnostic medical device’ means any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information on one or more of the following:
(a) concerning a physiological or pathological process or state;
(b) concerning congenital physical or mental impairments;
(c) concerning the predisposition to a medical condition or a disease;
(d) to determine the safety and compatibility with potential recipients;
(e) to predict treatment response or reactions;
(f) to define or monitoring therapeutic measures.
Specimen receptacles shall also be deemed to be in vitro diagnostic medical devices;
Section 1, Article 1, 3. Defines what the IVDR does not apply to:
products for general laboratory use or research-use only products, unless such products, in view of their characteristics, are specifically intended by their manufacturer to be used for in vitro diagnostic examinations;
invasive sampling products or products which are directly applied to the human body for the purpose of obtaining a specimen;
internationally certified reference materials;
materials used for external quality assessment schemes.
Fill out this form, or contact us at info@criterionedge.com, and we would be happy to answer any of your questions as well as book you an appointment to discuss your project needs during a time that works for you.
Are You Reg. Ready? Get your medical affairs team primed to manage new regulatory requirements
In this discussion, we provide an overview of how to plan and coordinate change within an organization to meet upcoming EU IVDR regulatory requirements. We will be highlighting key steps involved in helping medical affairs transition to a more rigorous regulatory environments, and how to evaluate talent gaps, team leadership composition, and process challenges of your product portfolio.
Driving Innovation to Success in the Market: Strategic Considerations
In this discussion, our panel of proven leaders discuss the key elements that support and propel the innovation process in the medical device, pharmaceutical, and IVD industries. We will be highlighting key areas of the process, important players in the pathway to the market, and how successful innovations spawn new innovations in new markets such as the digital health space, and more.
Privacy Overview
This website uses cookies so that we can provide you with the best user experience possible. Cookie information is stored in your browser and performs functions such as recognising you when you return to our website and helping our team to understand which sections of the website you find most interesting and useful.
Strictly Necessary Cookie should be enabled at all times so that we can save your preferences for cookie settings.
If you disable this cookie, we will not be able to save your preferences. This means that every time you visit this website you will need to enable or disable cookies again.
3rd Party Cookies
This website uses Google Tag Manager and Pardot's tracking features to collect information such as the number of visitors to the site, and the most popular pages. Keeping this cookie enabled helps us to improve our website.
Please enable Strictly Necessary Cookies first so that we can save your preferences!