The Food and Drug Administration (FDA) is promoting the Quality Overall Summary (QOS) as a powerful tool to promote effective communication between regulators and sponsors of drugs as well as a tool that can substantially impact the efficiency and quality of the regulator’s assessment. The QOS is required for all New Drug Applications (NDAs), Abbreviated New Drug Applications (ANDAs) and Biologics License Applications (BLAs), thus the QOS has significant potential to impact the regulatory review process for getting marketing approval.
The QOS summarizes all quality-related information in the application. As part of Module 2 of the electronic Common Technical Document (eCTD), the QOS links to the sponsor’s larger body of data in Module 3. The QOS is expected to provide the regulator with sufficient information to understand the contents of Module 3 in a high-level overview. However, FDA suggests that many sponsors are falling short of these expectations and are not fully utilizing this powerful tool as an effective guide for regulators to assess the application.
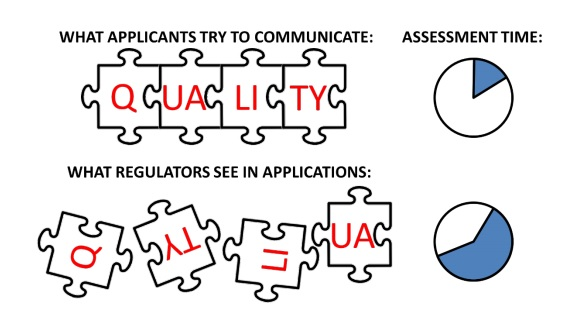
The QOS provides the sponsor with an opportunity to summarize the key aspects of the new drug or biologics application, explain specific items for the regulators to consider, and extend to post-approval comments. Yet, a poorly written QOS requires regulators to spend significant effort to “understand, summarize, collate, and interpret quality data from module 3 (Figure 1).
Figure 1. There can be a disconnect between applicants and regulators regarding the communication of quality data and its impact on the assessment. Currently, it takes time and/or communications (e.g., information requests) to fully understand the quality of data and its significance in an application.
The FDA’s white paper describes key considerations for creating a high–quality QOS to ensure regulators have a good idea of the potential risk to the patient and the control of this risk in the commercially manufactured product. The 3 key considerations are:
Biological products warrant special regulatory consideration because of their complex nature and susceptibility to variation during manufacturing. Biologics are not only complex in their physical structure, they are produced from living organisms and thus pose a myriad of potential issues in the manufacturing and isolation processes that all have the potential to induce immunogenicity. Regulations for developing a biological product take these potential risks into consideration
In this second piece evaluating BLA and NDA, we focus on understanding some of the nuances between biologic and drug development. See the first BLA vs NDA blog for a more focused look at regulations.
Manufacturing of Biological Products is Inherently Riskier than Production of Drugs.
The manufacturing processes for biological products are different than processes for pharmaceuticals. Traditional drug products are typically manufactured using pure chemical substances that are sterile, and the end products can be relatively easily analyzed. On the other hand, biological products are made from living organisms and are much more complex in nature — making product analysis very difficult. Indeed, most biological products are defined by the manufacturing processes used for production. The manufacturing process and manufacturing facilities are so crucial to biologics that “purity” is part of the agency’s requirements for licensing.
The unique characteristics and manufacturing processes of therapeutic
biological products and drug compounds lays the framework for the differences
in regulatory requirements for getting into the marketplace. While, biologics and drugs are both used for the same purposes — to
treat, prevent, and cure diseases — biological products are much more complex
in nature. By comparison, common drug compounds are relatively simple.
What exactly is a biological product?
Biological products are comprised of large and
complex protein structures that are primarily derived from living material,
including human, animal, and microorganisms. Proteins are often
post-transcriptional modified, including glycosylation, oxidation, deamidation,
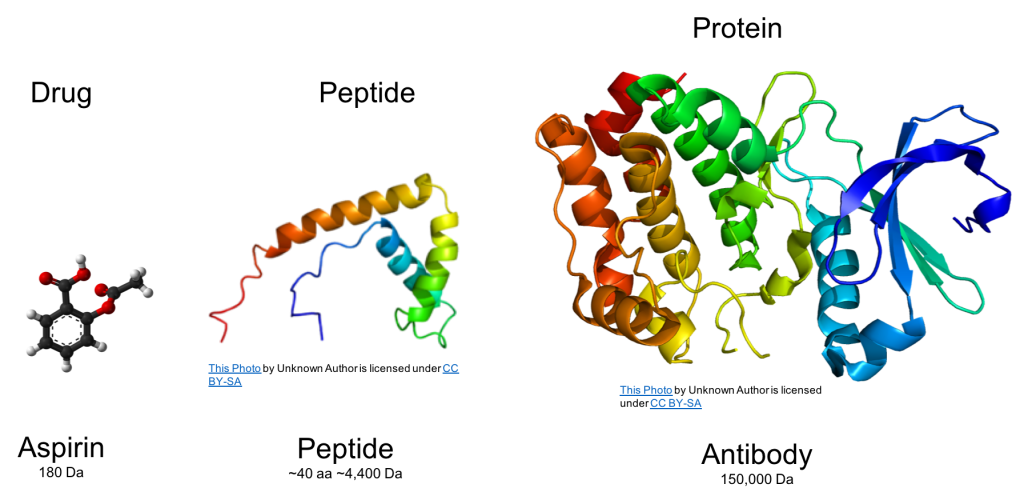
and this has a profound effect on protein properties. As seen in the figure below, this contrast
with conventional drug compounds, such as aspirin, that have a smaller
molecular weight and are chemically synthesized. Peptides can fall into either regulatory
category and are comprised of amino acids just like a protein, but peptides are
smaller.
The vast
differences in complexity and size are depicted in this figure.
Defining biological products and drug compounds is the
first step to understanding the common and unique regulatory requirements for
each. FDA’s definition is the only one that matters for the purpose of
obtaining marketing approval in the United States, and the definition for
biologics is in a transition period.
Fill out this form, or contact us at info@criterionedge.com, and we would be happy to answer any of your questions as well as book you an appointment to discuss your project needs during a time that works for you.
Are You Reg. Ready? Get your medical affairs team primed to manage new regulatory requirements
In this discussion, we provide an overview of how to plan and coordinate change within an organization to meet upcoming EU IVDR regulatory requirements. We will be highlighting key steps involved in helping medical affairs transition to a more rigorous regulatory environments, and how to evaluate talent gaps, team leadership composition, and process challenges of your product portfolio.
Driving Innovation to Success in the Market: Strategic Considerations
In this discussion, our panel of proven leaders discuss the key elements that support and propel the innovation process in the medical device, pharmaceutical, and IVD industries. We will be highlighting key areas of the process, important players in the pathway to the market, and how successful innovations spawn new innovations in new markets such as the digital health space, and more.
Privacy Overview
This website uses cookies so that we can provide you with the best user experience possible. Cookie information is stored in your browser and performs functions such as recognising you when you return to our website and helping our team to understand which sections of the website you find most interesting and useful.
Strictly Necessary Cookie should be enabled at all times so that we can save your preferences for cookie settings.
If you disable this cookie, we will not be able to save your preferences. This means that every time you visit this website you will need to enable or disable cookies again.
3rd Party Cookies
This website uses Google Tag Manager and Pardot's tracking features to collect information such as the number of visitors to the site, and the most popular pages. Keeping this cookie enabled helps us to improve our website.
Please enable Strictly Necessary Cookies first so that we can save your preferences!